Usage and example:
Workflow of pre-processing:
Import packages:
import scanpy as sc
import numpy as np
import giniclust3 as gc
import anndata
Import single cell count matrix from csv file:
adataRaw=sc.read_csv("giniclust3/data/GSM1599495_ES_d0_biorep_techrep1.csv",first_column_names=True)
Filter gene expression matrix:
sc.pp.filter_cells(adataRaw,min_genes=3)
sc.pp.filter_genes(adataRaw,min_cells=200)
Transform expression matrix (skip this step if the input matrix is: col for genes and row for cells):
adataSC=anndata.AnnData(X=adataRaw.X.T,obs=adataRaw.var,var=adataRaw.obs)
Gene expression normalization:
sc.pp.normalize_per_cell(adataSC, counts_per_cell_after=1e4)
Apply GiniClust3 for both common and rare cluster identification:
Perform GiniIndexClust:
gc.gini.calGini(adataSC) ###Calculate Gini Index
adataGini=gc.gini.clusterGini(adataSC,neighbors=3) ###Use higher value of neighbor in larger dataset. Recommend (5:15)
Perform FanoFactorClust:
gc.fano.calFano(adataSC) ###Calculate Fano factor
adataFano=gc.fano.clusterFano(adataSC) ###Cluster based on Fano factor
Generate conseneus matrix and apply ConsensusClust step:
consensusCluster={}
consensusCluster['giniCluster']=np.array(adataSC.obs['rare'].values.tolist())
consensusCluster['fanoCluster']=np.array(adataSC.obs['fano'].values.tolist())
gc.consensus.generateMtilde(consensusCluster) ###Generate consensus matrix
gc.consensus.clusterMtilde(consensusCluster) ###Cluster consensus matrix
####output cluster results to final.txt####
np.savetxt("final.txt",consensusCluster['finalCluster'], delimiter="\t",fmt='%s')
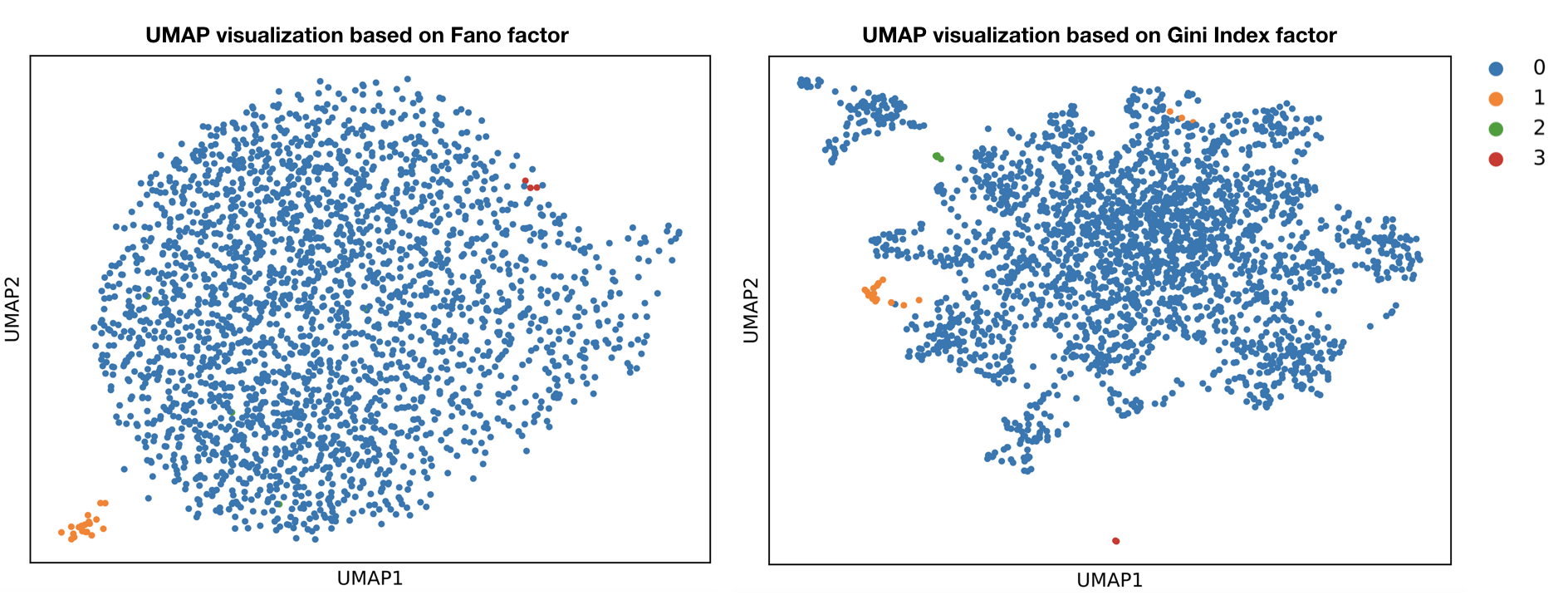
UMAP visualization:
adataGini.obs['final']=consensusCluster['finalCluster']
adataFano.obs['final']=consensusCluster['finalCluster']
gc.plot.plotGini(adataGini)
gc.plot.plotFano(adataFano)
UMAP visualization of the figures